The Food and Drug Administration (FDA) has released their long-awaited proposed rule on prescription drug importation. President Trump has mentioned his plan at events in recent months, citing the plan as part of the administration’s efforts to lower prescription drug costs. The proposed rule outlines requirements for so-called Section 804 Importation Programs (SIPs), named after the section of the Federal Food, Drug, and Cosmetic Act (FD&C Act) under which the statutory authority comes from. States will be permitted to develop these importation programs, identifying drugs, sellers, and importers who may participate. Comments on the proposed rule are due March 9, 2020.
The rule was released in conjunction with draft guidance for prescription drug manufacturers that establishes steps to be taken by manufacturers for their drugs that would be eligible for importation. Most notably, a manufacturer would be required to file a supplement to the drug’s New Drug Application (NDA) before the product would be eligible for importation. Because there is nothing in the law that compels a manufacturer to do this, a drug product would not be eligible for importation without the manufacturer’s explicit consent. Comments on the draft guidance are due March 5, 2020.
Finally, the FDA is also seeking comments on broader issues related to the potential impact of importation on labeling, prescription drug safety, and the biosimilar/generic drug market. Those comments are due Friday, February 21, 2020.
FDA to Authorize Section 804 Importation Programs
Under statute, the importation of prescription drugs from Canada is allowed under certain conditions if certified by the Secretary of the Department of Health and Human Services (HHS). The Secretary must certify that importation will “pose not additional risk to the public’s health and safety” and “result in significant reduction in the cost of covered products to the American consumer.” With this proposed rule, the administration is proposing to allow time-limited Section 804 Importation Programs (SIPs), which would be authorized by the FDA and managed by states or certain other entities.
All SIPs would be sponsored by a state, tribal, or territorial government, but may have cosponsors. These co-sponsors, referred to as SIP Sponsors, could be a pharmacist, wholesaler, or other State or non-federal government entity. The FDA believes that allowing co-sponsors may increase flexibility, but the agency is seeking comments on this, particularly comments on what the division of responsibility between co-sponsors should be, whether certain arrangements should not be allowed, and an alternative option to allow pharmacists or wholesalers to sponsor a SIP without a co-sponsor.
Importation of Biologics, Controlled Substances, and Drugs that are Infused, Injected, or Subject to REMS Would be Prohibited
A sponsor would have to specify the following in a SIP proposal:
- Eligible prescription drugs: these are the drugs that would be included in a SIP and must be approved for sale in either the Canadian or American market;
- Foreign seller: entity in Canada that will purchase to drug directly from its manufacturer and must be licensed by Health Canada as a wholesaler and registered with the FDA; and
- Importer: entity in the United States that will buy the drug directly from the foreign seller and must be a wholesaler or pharmacist licensed in the U.S.
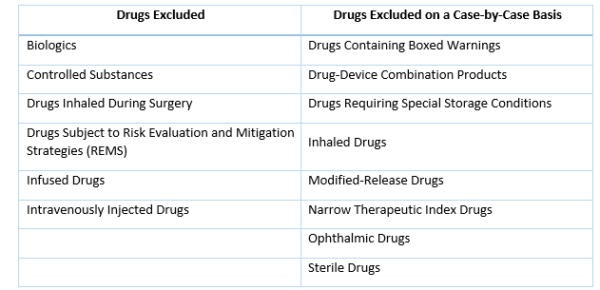
Several categories of drugs are excluded from eligible prescription drugs based on statute, and the FDA is proposing to only allow importation of a second set of drugs on a case-by-case basis:
A SIP proposal will also need to show that the importation will pose no additional risk to public health and safety and explain why this would result in a significant cost reduction to consumers using the imported drug. A variety of information must be in the SIP proposal, including where the active ingredient for each drug is manufactured; the FDA-registered re-packager or re-labeler in the United States; and how the SIP sponsor will educate pharmacists, healthcare providers, and patients about the SIP among others.
Initially, SIPs will identify one foreign seller and one importer; a supplemental proposal to add additional sellers or importers may be submitted once the SIP demonstrates it can consistently import eligible drugs. Only three entities may be in the supply chain for each drug: one manufacturer, one foreign seller, and one importer.
The FDA would authorize SIPs for two-year periods. SIPs may be extended by the FDA prior to the end of its approval period; extensions would be for an addition two years. The FDA can choose not to authorize a SIP proposal and the FDA may also terminate a SIP at any time for reasons such as safety concerns, likelihood the SIP may not result in significant enough cost savings, or because the FDA needs to limit the number of authorized SIPs to efficiently run the program.
Manufacturers Would Have to Submit a Supplemental NDA Prior to Importation
In the draft guidance released concurrently with the proposed rule, the FDA states that a manufacturer would be required to submit a supplemental new drug application (NDA) before a product would be eligible for importation. This raises a question of how impactful the overall program will be, as manufacturers could potentially refuse to file a supplemental NDA, therefore making their products ineligible for importation.
Separate NDC Numbers May Alleviate Pricing Concerns
According to the draft guidance, after approval of a supplemental NDA, the manufacturer would receive a new National Drug Code (NDC) number for the FDA-approved imported prescription drug. Manufacturers would also have to ensure that imported drug labels reflect the U.S. marketed name and safety information, if there are differences.
The ability for manufacturers to assign a unique NDC number to imported products will also allow use of differential pricing between imported and non-imported drugs. Because manufacturers calculate and report both average sales price (ASP) and average manufacturer price (AMP) using NDC numbers, manufacturers, if they wish, will be able to establish a pricing strategy for imported drugs that is independent of their pricing strategy for non-imported drugs. The FDA hopes different NDC numbers will also support pharmacovigilance, accurate billing and reimbursement, and facilitate clearance of the imported drugs through customs.
Importers Must Follow Pre-Import, Labeling, and Testing Requirements
Under an authorized SIP, the importer would submit a pre-import request to the FDA at least 30 days before the scheduled date of arrival or entry for consumption of the drug shipment, whichever is earlier. Among the items included on the pre-import request is a detailed description of a testing plan. All shipments of imported drugs would have to arrive through a U.S. Customs and Border Protection (CBP) port of entry authorized by the FDA and the importer would have to file appropriate documentation. Both the foreign seller and the importer would be subject to supply chain security requirements, including Drug Supply Chain Security Act (DSCSA) obligations.
Each package of imported drugs would have to be labeled with a section 804 serial identifier (SSI) and a National Drug Code (NDC) number. An SSI will be an alphanumeric serial number that is unique to each package or homogenous case. The importer, in addition to this labeling, will need to maintain records related to this.
The drug importer will also have to arrange for testing of the drug for authenticity, degradation, and other statutory testing requirements by a qualifying laboratory in the United States if the manufacturer does not perform the required testing. Information about the testing would be included in the SIP proposal. Test results would be reviewed and accepted by the FDA and then the drug would have to be re-labeled to align with statutory labeling requirements under the FD&C act before being distributed.
FDA Outlines Post-Importation Requirements
The proposed rule also includes post-importation requirements. SIP Sponsors would be required to provide the FDA with various data and information about the SIP, including the cost savings for consumers. Adverse event, medication error, field alert, and other reports would have to be submitted by the importer to the FDA and the drug’s manufacturer. Should it be necessary, a SIP sponsor would also be responsible for implementing a recall. The rule also proposes that SIP sponsors have a written recall plan in place that includes procedures to perform a recall and who would be responsible for performing such procedures.
FDA Declines to Predict Financial Impact of Proposed Rule
The proposed rule does not predict or estimate how this rule may affect the U.S. market for prescription drugs. The agency says it lacks the information about the expected scale or scope of such importation programs so it is unable to estimate its impact on the market, particularly related to the volume or value of drugs that may be imported and the savings to consumers.
The proposed rule does specifically note that it does not intend to address the applicability of the Medicaid drug rebate program for drugs in a SIP and further mentions that this issue may be addressed in future guidance or rulemaking from HHS if appropriate.
FDA Separately Seeks Comment on Broader Issues Related to Labeling, Impact of Importation on Biosimilar/Generic Market, and Safety Concerns
The agency is also seeking feedback on more general questions, such as how importation might impact generic or biosimilar drug development and market entry. Specifically, the FDA is interested in the following feedback:
- How the proposed labeling guidance could be applied, or may need to be adjusted, to account for certain types of drug products, including (but not limited to) sterile injectables, drugs with boxed warnings, drugs subject to a Risk Evaluation and Mitigation Strategy (REMS), controlled substances, and combination products.
- Phrasing recommendations for a labeling statement that identifies the products as imported, or whether there are other distinguishing characteristics or communication methods (e.g. a Dear Provider or Pharmacist Letter) that would be equally effective.
- How much it would cost a manufacturer, repackager, or relabeler to implement the alternative labeling and any alternative approaches.
- Whether manufacturers of multiple-source drugs (those approved under an Abbreviated New Drug Application) would also be in need of additional NDC numbers, especially with regards to pricing.
- Potential impact of importation on biosimilar and generic drug development and market entry.
- Potential safety risks related to importation that could be addressed via regulation.
The FDA will accept comments on these issues through Friday, February 21.